


















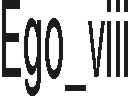










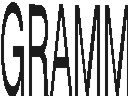






































The Linux for Chemistry project (LfC) is dedicated to compiling the most comprehensive collection of Chemistry software, and making it available for the Linux operating system. LfC provides point-and-click installation, a complete on-line library of searchable documentation, and much more.
The packages on LfC represent the state-of-the-art in Chemistry data processing, and are identical to the versions used on high end scientific workstations.
The combination of ever decreasing hardware prices, and the superb Linux operating system, now makes it possible to use these same packages on a "standard" personal computer. The current LfC distribution contains Linux versions of the following Chemistry data reduction and analysis packages :
|
| 2dhf | 2 Dimensional Finite Difference Hartree-Fock Program (WWW) | ||
|
| ASICS | Analysis of single crystal spectra (WWW) | ||
|
| ARTwork | A ma"center"> | ARTwork | A materials simulation tool. (WWW) |
|
| atomc | Monte Carlo simulation of atomic fluids in the (T,V,N), (T,P,N), or (T,V,mu) ensemble NB. This program is distributed as an encrypted archive, please email Mihaly Mezei to request the decryption key. (WWW) | ||
|
| Babel | A Molecular Structure Information Interchange Hub (WWW) | ||
|
| big_mac | Configurational Bias Monte Carlo code (WWW) | ||
|
| Ballroom | A Molecular animation viewer (WWW) | ||
|
| Boost | C++ programming libraries (WWW) | ||
|
| BSxbs | ball-and-sticks plotting program (WWW) | ||
|
| ccsl | "ccsl.gif" ALIGN="center"> | ccsl | The Cambridge Crystallography Subroutine Library (WWW) |
|
| CDA | Charge Decomposition Analysis (WWW) | ||
|
| chemeq | Chemical equilibrium calculator | ||
|
| Chemtool | Molecular drawing program (WWW) | ||
|
| Chooch | Calculation of Anomalous scattering factors from X-ray fluorescence data (WWW) | ||
|
| Cn3D | NCBI structure viewer (WWW) | ||
|
| CRK | Chemical resource kit (WWW) | ||
|
| dacapo | Ab initio molecular dynamics code, based on ultra-soft pseudopotentials. (WWW) | ||
|
| Dino | Visualizing Structural Biology (WWW) | ||
|
| DRAWxtl | Program to read crystallographic information such as unit cell, space group, atom positions, and thermal ellipsoid parameters to generate POLYHEDRON, SPHERE, ELLIPSOID and BOND geometry POV and VRML scenes. (WWW) | ||
|
| discus | Simulate crystal structures and calculate the corresponding Fourier transform (WWW) | ||
|
| dnd | Molecular Visualization (WWW) | ||
|
| ego_viii | A program to compute molecular dynamics trajectories. (WWW) | ||
|
| elf | A script for preparing ELF (Electron Localisation Function) files, using Gaussian's Wavefunction output. (WWW) | ||
|
| equiv | A program for the analysis and management of data collections from X-ray single crystal diffractometry (WWW) | ||
|
| fhi96md | molecular dynamics simulations and total-energy calculations (WWW) | ||
|
| Fhkl | Computation of crystal characteristics (WWW) | ||
|
| Formulae | Formulae of common chemicals (WWW) | ||
|
| fungimol | Fungimol is an extensible system for designing atomic-scale objects (WWW) | ||
|
| gamma | A General Approach To Magnetic Resonance Mathematical Analysis (WWW) | ||
|
| g3data | g3data is used for extracting data from graphs. In publications graphs often are included, but the actual data is missien are included, but the actual data is missing. g3data makes the extracting process much easier. (WWW) | ||
|
| geomview | 3d viewer (WWW) | ||
|
| gdpc | gdpc is a program for visualising molecular dynamic simulations (WWW) | ||
|
| gibbsG | ibbs-ensemble simulation with cavity-biased insertion NB. This program is distributed as an encrypted archive, please email Mihaly Mezei to request the decryption key. (WWW) | ||
|
| GraphEnt | Graphical Maximum Entropy modeller (WWW) | ||
|
| gperiodic | Periodic table browser (WWW) | ||
|
| gramm | GRAMM is a program for protein docking (WWW) | ||
|
| intocham, | conversion from InsightII to Charm, Amber, or Moil, NB. This program is distributed as an encrypted archive please email Mihaly Mezei to request the decryption key. (WWW) | ||
|
| IMC | Iterative Mobile ClusteringMultiple-Site Ligand Binding to Flexible Macromolecules (WWW) | ||
|
| JChemPaint | JChemPaint is a Java 2 program for drawing 2D chemical structures (WWW) | ||
|
| jana2000 | Chrystallographic computing system (WWW) | ||
|
| kuplot | General purpose plotter (WWW) | ||
|
| laue | XLaue diagrams (WWW) | ||
|
| lennard | Lneeard-Jones simulation | ||
|
| LOOPP | Learning, Observing and Outputting Protein Patterns (WWW) | ||
|
| maxwell | multipole expansion for condensed phases NB. This program is distributed as an encrypted archive please email Mihaly Mezei to request the decryption key. (WWW) | ||
|
| mmc | Monte Carlo program NB. This program is distributed as an encrypted archive please email Mihaly Mezei to request the decryption key. (WWW) | ||
|
| MEAD | Macroscopic Electrostatics with Atomic Detail (WWW) | ||
|
| MOIL | The MOIL modeling package simulates structure, dynamics, and function of biological molecules at the atomic level of detail. (WWW) | ||
|
| moldy | Molecular dynamics simulations (WWW) | ||
|
| MOPAC7 | Semi-empirical molecular orbital package (WWW) | ||
|
| moscito | Molecular dynamics package (WWW) | ||
|
| MPQC | Massively parallel quantum chemistry toolkit (WWW) | ||
|
| MMTK | Molecular modelling toolkit (Python) (WWW) | ||
|
| modelfree | A program for optimizing "Lipari-Szabo model free" parameters to heteronuclear relaxation data (WWW | ||
|
| MosView | Viewer for moscito simulations (WWW) | ||
|
| NAB | Nucleic Acid Builder (WWW) | ||
|
| nmrview | Visualization and analysis of NMR datasets. (WWW) | ||
|
| ORAC | A molecular dynamics program to simulate solvated biomolecules (WWW) | ||
|
| ORTEP | Oak Ridge Thermal Ellipsoid Plot Program for Crystal Structure Illustrations (WWW) | ||
|
| PASTA | PASTA Toolkit, Protein ASsignment by Threshold Accepting (WWW) | ||
|
| pdffit | Full profile structural least square refinement of the atomic pair distribution function (WWW) | ||
|
| pepinsky | Pepinsky's Machine : an interactive, graphics-based Fourier synthesis program with applications in teaching and research (WWW) | ||
|
| ppovit | POV-Ray molecular raytracing (WWW) | ||
|
| PyMol | Molucular Visualization toolkit (WWW) | ||
|
| Qs | Queen of Spades. a n-dimensional, space-group general, molecular (re)placement program (WWW) | ||
|
| RasMol | Molecular graphics Visualization tool (WWW) | ||
|
| Raster3D | 3D Molecular graphics rendering tool (WWW) | ||
|
| Raster3D | 3D Molecular graphics rendering tool (WWW) | ||
|
| Scientific Python | A collection of Python modules that are useful for scientific computing (WWW) | ||
|
| simloc | detection of local similarities NB. This program is distributed as an encrypted archive please email Mihaly Mezei to request the decryption key. (WWW) | ||
|
| Simpson | Solid-state NMR simulation program. (WWW) | ||
|
| SIMREF | Simultaneous Rietveld RTD> | Simultaneous Rietveld Refinement with Multiple Powder Data Sets (WWW) | |
|
| simulaid | simulation setup utilites NB. This program is distributed as an encrypted archive please email Mihaly Mezei to request the decryption key. (WWW) | ||
|
| Situs | A package for the docking of atomic resolution structures to low-resolution density maps (WWW) | ||
|
| SPDBV | Swiss PDB viewer (WWW) | ||
|
| SSIA | SSIA is a program for predicting the magnitude and orientation of a sterically induced alignment tensor from a solute's three-dimensional shape (WWW) | ||
|
| steric | A Program to Calculate the Steric Size of Molecules about a Point in terms of their Cone Angles and Solid Angles, as well as their total Volumes and Projected Areas (WWW) | ||
|
| vaspview | Scientific visualization package for examining output files generated by the Vienna Ab-initio Simulation Package (WWW) | ||
|
| tessel2 | A 3D "compiler" to produce crystal and molecular models, c parametric surfaces and several forms of sphere tesselations. (WWW) | ||
|
| vievmol | Viewmol is an open source graphical front end for computational chemistry programs (WWW) | ||
|
| vis5d | 3D viewer (WWW) | ||
|
| XCombust | Elemental analysis (WWW) | ||
|
| WMOVIEC | Wpg" ALIGN="center"> | WMOVIEC | WMOVIEC is a program that creates movies of atomic motion. (WWW) |
|
| XITE | X-based Image Processing Tools and Environment (WWW) | ||
|
| XMolCalc | Molecular weight calculator (WWW) | ||
|
| Xmakemol | A program for viewing and manipulating atomic/molecular systems (WWW) | ||
|
| xnmr | Processing and simulation of exchange broadened NMR spectra SHAREWARE (WWW) | ||
|
| xtal | Xtal3.7 System of Crystallographic Programs (WWW) |
LfC also now includes a set of graphical and database tools, tools for enhancing the security of your Linux system, and a remote desktop package (VNC).
All the applications have been tested with the Linux system distributions from RedHat (version 7.2) and SuSE (version 7.1). (a full listing of contents of the current release is available here, please note that this is a large file download.)
The collection has been assembled with the assistance of the worldwide Chemistry software community. The system includes an integrated set of documentation in HTML format, and many packages include PostScript™ format documents. Source code is included where permitted by the package license holders.